

FREQUENTLY ASKED QUESTIONS & ISSUES
Below are listed some of the most frequently asked questions, covering General, RAMP IRB-related and Specific FAQs. To find an answer, click on an FAQ. To return to the list, click again on the FAQ or "v" symbol. For FAQs specific to student-led research, visit this student-led research FAQs page.
Search FSU
- RAMP IRB is accessible within the myFSU portal as an icon under the myFSU Links section, and you must therefore have a valid FSU user credential. The system is designed to be part of the single sign-on process, allowing FSU credentialed users to conveniently access RAMP IRB along with other systems within a single platform using one set of login credentials. Use any of the means below to access the RAMP IRB system to submit your study:
- Sign on to the myFSU portal https://www.my.fsu.edu/portal and click on the RAMP icon;

- Visit https://myramp.research.fsu.edu; or,
- Click on the link from an auto-generated email from ramp-irb@fsu.edu
- If you are experiencing technical issues accessing RAMP, try clearing your browsers' history and/or use another internet browser, then try logging into RAMP again. If the issues continue, contact the FSU RAMP Support Office at RAMP-Grants@fsu.edu or dmullins2@fsu.edu.
- Sign on to the myFSU portal https://www.my.fsu.edu/portal and click on the RAMP icon;
- Non-FSU researchers are generally not provided with access to any RAMP module, including RAMP IRB. However, in very limited circumstances, non-FSU individuals who are collaborating researchers may request Sponsored Guest Access. Such requests must first be formally approved by Department Chair, Associate Dean or designee, and then cleared by OHSP/IRB. All requests for a Sponsored Guest access must be submitted to the RAMP-Grants@fsu.edu mailbox with the subject: Sponsored Guest Access Request (see this RAMP Support Office link for additional information and instructions). The OHSP/IRB will not clear such requests unless the FSU PI has already submitted an IRB application for review, and the application includes sufficient information about the non-FSU researcher(s).
- IMPORTANT NOTE: The RAMP IRB SmartForm application uses business logic to guide you in completion of your submission. The Smartform application also includes on most pages and sections many ? (or Help) icons and buttons to provide you with key instructions and information about what is required to be answered, uploaded or provided for purposes of review, and where to obtain templates and forms. Save time and effort and click on these ? icons to get answers and directions! See sample images below:


- Video tutorials are available to assist you with using RAMP IRB and navigating a study submission workspace. Tutorials last just a few minutes and include video captures of RAMP IRB workspaces. To access the tutorials go here.
- When study documents (e.g., study protocol, consent forms) must be submitted, many templates are available, each tailored to the scope and complexity of your research; these are found under the IRB, Library and Templates tabs. Note that when using protocol templates, some sections may not be applicable to your research; if so you may mark as “NA”, but do not delete the section, otherwise your protocol will be returned to you for correction.
- For step-by-step instructions, refer to the Researcher's Guide to RAMP IRB, or to the OHSP Researchers Training slides available in RAMP IRB Help Center.
- For other questions about using RAMP IRB, including obtaining assistance with using RAMP IRB, visit this general RAMP IRB FAQ web page, also available at: https://ramp.research.fsu.edu/faqs/irb/
- Respond to a Clarification Request in RAMP IRB, not by email or telephone call since these responses cannot be entered by staff into the RAMP regulatory file for your study. In the RAMP IRB submission workspace for the study, under the “History” tab, carefully review the requested clarification(s). When you are ready to provide the requested information and documents, under Next Steps click “Edit Study”, make any requested changes to the SmartForm and/or upload requested documents, and then click “Exit” on the SmartForm once all revisions have been completed. Be certain to click “Submit Response” on the study workspace (to the left of the workflow diagram) and then click OK. Only then will your submission return to our queue for further handling.
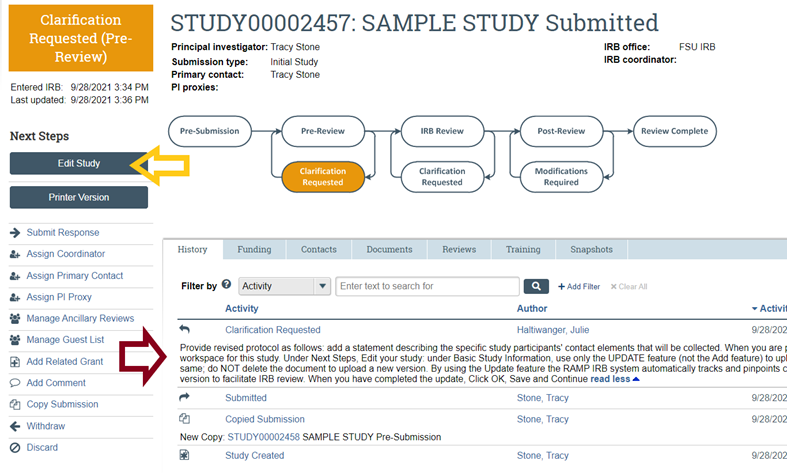
- Do not attach study-related documents to a comment or response submission since these materials cannot be readily accessed or finalized by the IRB or maintained with other study documents when the documents are located outside of the proper submission or SmartForm location or section.
- In limited circumstances and only as an interim measure you may be instructed to provide the IRB with requested information or study-related documents as an attachment to a comment or response submission; note however that you will still be required to provide those documents in the correct workspace or SmartForm location when your study is made available for you to edit so that these materials may be finalized by the IRB.
- To submit a Continuing Review, log into RAMP IRB and under the IRB and Help Center tabs refer to the IRB Researcher's Guide on p. 11 for instructions on creating and submitting a Continuing Review to renew your IRB approval before IRB approval expires.
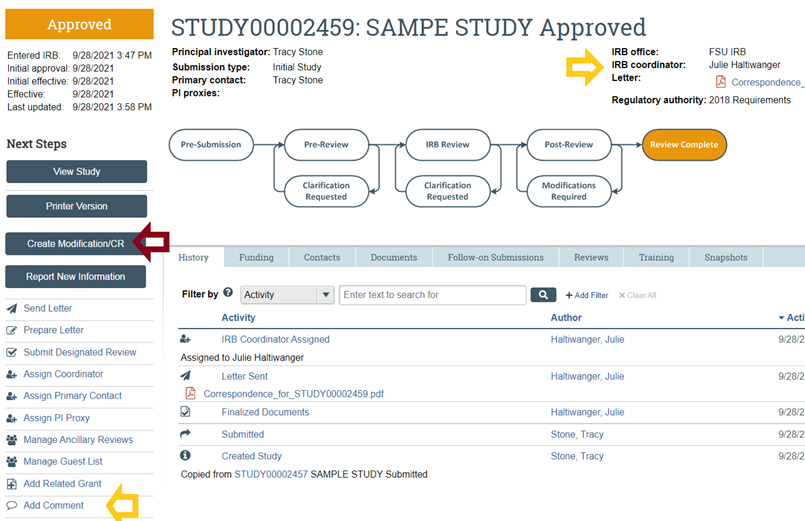
- If you have additional questions use the “Add Comment” feature (to the left of the workflow diagram and listed below under Next Steps) on your study workspace to ask your question and select the option to send an email notification about your question to your IRB Coordinator (whose name is listed above and to the left of the workflow).
- To make a change to or revise your study: log into RAMP IRB and under the IRB and Help Center tabs refer to the to the IRB Researcher's Guide on p. 11, then follow the instructions on submitting a Modification. Note the modification scope “Other parts of the study” will allow you to edit any of the original SmartForm sections except for study team member information; if you need to make changes to both study team member information and another section of the SmartForm, then both scopes should be selected.
- Was your study determined to be exempt from IRB review? If so, then in the official exemption determination letter that you received for this study you will find a list of planned study changes that do not and do require submission for review. Look over these examples to see if your proposed changes will require review. If so or if you are not sure, follow the instructions on p. 11 of the IRB Researcher's Guide to create and submit a modification for this study.
- To close out your study: log into RAMP IRB and from the Help Center refer to the IRB Researcher's Guide on p. 11, then follow the instructions for submitting a Continuing Review (which process is also used to close a study). Note that in order to close a study, the first 4 research milestones in section 4 on the Continuing Review/Study Closure page must be checked. BE sure to acknowledge that the study will be closed, discard any open follow-on submissions related to the study, complete any remaining items or respond to any other questions, attach any supporting documents as may be needed, then select Save, Finish and under Next Steps select Submit. Your Study Closure submission will move into the queue for review and further handling.
- If you have additional questions use the “Add Comment” feature (to the left of the workflow diagram and listed below under Next Steps) on your study workspace to ask your question and select the option to send an email notification about your question to your IRB Coordinator (whose name is listed above and to the left of the workflow).
FSU researchers:
- CITI training completion documentation is synced with RAMP IRB each evening through a nightly data transfer. You should wait for the next day after CITI training course completion to add yourself to a RAMP IRB submission.
![]()
- Your CITI profile must:
- Affiliate with FSU, and
- Be accurate, including using your official FSU email address as the primary email.
- The data transfer feature is a courtesy to the FSU research community, and made possible by separate arrangement between the 2 proprietary vendors that support CITI and RAMP IRB. However, if your CITI training completion documentation does not transfer, then you will be required to upload the CITI training completion documentation within the RAMP IRB submission workspace.
- Refer to the Investigator Manual for information about CITI human subjects research training requirements or visit our website’s training page for step-by-step instructions on how to enroll in the correct training: https://www.research.fsu.edu/research-offices/ohsp/investigator-resources/citi-training-requirements/. Only the specified training will suffice for IRB review purposes.
Non-FSU researchers:
- You must provide the FSU Principal Investigator with a copy of your CITI training completion documentation, and the FSU Principal Investigator must upload the copy in other study documents within the RAMP IRB submission workspace.
- Always log in to RAMP IRB and view your study submission’s workspace to check the status of your submission. The workflow diagram and the History tab entries will indicate your study’s submission status. In the figure below, the example workflow diagram indicates that your study is in the Pre-Submission state (the study has not yet been submitted to OHSP and IRB for their review), and under the History tab it shows that you have created (but not submitted) your study.
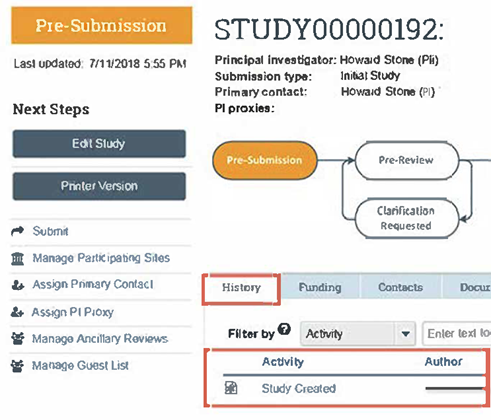
- For a more detailed explanation about using the workflow diagram to check the status of your submission, click on this RAMP IRB workflow explainer.
- Be sure to follow instructions on the Final Page in the RAMP IRB SmartForm for your study, which states that you must “click Submit” in order for your study to ever enter the IRB review queue. With many thousands of FSU researchers, the IRB does not search out for studies in the Pre-Submission stage to follow up for reasons in delay as there may be a myriad of reasons why study staff maintain their studies in the Pre-Submission.
- Note that only these individuals who are authorized to actually submit to the IRB (i.e., Principal Investigators (PIs) or PI proxies) will be able to see the “Submit” button. If you are not the PI or PI Proxy for the study, then you will not be able to see nor execute the “Submit” feature. The study PI or PI Proxy needs to log in; they will see the “Submit” button above the “Assign Primary Contact” button. The PI has the option to “Assign PI Proxy” so therefore they can click Submit or they assign another study team member as PI Proxy; that study team member will then have the authority to click “Submit” on the PI’s behalf for this study.
- Special note if you receive the notification “Response Time Exceeded”: these notifications are only intended as a courtesy to alert you that it has been 14 days since clarification was requested on your submission and our office has not yet received your response. You may still make or submit edits and take the time that you need to do (unless you have been specifically told about a deadline for re-submission; check your correspondence from IRB). You will continue to receive these notifications until you take the required action noted in the Clarification Requested instructions.
- Refer to our Human Research Review web page to learn about IRB turn-around times. The referenced anticipated turn-around times apply only to submissions that are complete (required forms, materials, information and requested clarifications have been submitted or completed, including completion of the required CITI human subjects training for all study staff). Carefully review ALL requirements before submitting otherwise you will risk needless further delay that could have been anticipated and avoided.
- If you have about the status of your submission, contact OHSP use the “Add Comment” feature (to the left of the workflow diagram and listed below under Next Steps) on your study workspace to ask your question and select the option to send an email notification about your question to your IRB Coordinator (whose name is listed above and to the left of the workflow); there, in response to the question about who should receive the email notification about your comment, select "IRB Coordinator." This will help to ensure that the IRB Coordinator assigned to your submission will receive a notification that your comment has been posted in your RAMP IRB workspace. Alternatively and if you have not yet submitted your study (i.e., your study is still in the Pre-Submission status) or an IRB Coordinator has not yet been assigned to your submission (i.e., no name is listed next to "IRB Coordinator"), contact OHSP or refer to the contact information in the left panel, and your question will be routed to OHSP staff for follow-up with you directly.
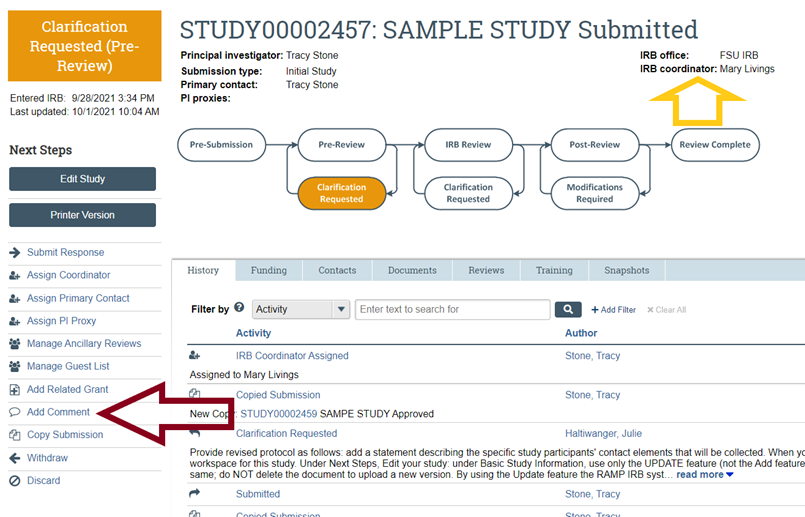
- If you have a RAMP IRB-related question not listed above, contact OHSP using the “Add Comment” feature (to the left of the workflow diagram and listed below under Next Steps) on your study workspace to ask your question and select the option to send an email notification about your question to your IRB Coordinator (whose name is listed above and to the left of the workflow); there, in response to the question about who should receive the email notification about your comment, select "IRB Coordinator." This will help to ensure that the IRB Coordinator assigned to your submission will receive a notification that your comment has been posted in your RAMP IRB workspace.
- Alternatively and if you have not yet submitted your study, contact OHSP or refer to the contact information in the left panel, and your question will be routed to OHSP staff for follow-up with you directly.
- For RAMP IRB-related FAQs specific to student-led research, visit this student-led research FAQs page.
- Many incidents (e.g., adverse events, breach of confidentiality, awareness of abuse/neglect/misconduct, protocol deviations) require reporting to the IRB within business 5 days from when researchers become aware of the incident. Refer to page 3 on our HRP-214 - Reportable New Information form to see what incidents require reporting; refer also to the section, "What are my obligations after IRB approval?" in the Investigator Manual.
- To submit a Report of New Information: log into RAMP IRB and under the IRB and Help Center tabs refer to the IRB Researcher's Guide on p. 13, then follow the instructions on creating and submitting a Reportable New Information.
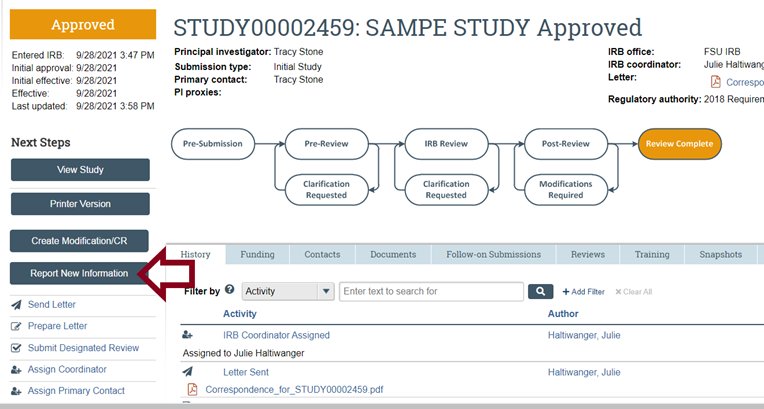
- Only if RAMP IRB is not accessible because of scheduled downtime or other circumstances and due to which you may miss the required reporting to the IRB within 5 business days, you may complete and send by email (humansubjects@fsu.edu) attachment a Report of New Information (RNI) (HRP-214 - FORM - Reportable New Information). The RNI will be acknowledged and reviewed and, as needed, you will be contacted for further information and instructions (which may include resubmitting the RNI through RAMP IRB once accessible).
Yes, a CAMS COI submission will affect review of an IRB submission in one or more of the following ways:
- Any RAMP IRB submission involving FSU employees or agents may result in a separate CAMS system notification to the employee or agent of their need to satisfy a CAMS disclosure and/or reporting requirement. If so, the employee or agent should take prompt action in CAMS to satisfy these requirements in order for a RAMP IRB submission to proceed to human research regulatory review.
- Note that each RAMP IRB submission for a new study, or a study modification involving addition of study staff for an active study, may require a separate CAMS disclosure and/or reporting for each study staff. This is not intended to be duplicative: applicable law requires that each FSU employee or agent's disclosure or reporting of financial or other interests be separately evaluated for its relation to and affect upon the design, conduct and reporting for each study (not all studies collectively) in which they may be involved, as an interest's relation or affect may differ among studies (e.g., a researcher's disclosure or report of a financial interest related to or possibly affecting one study may be unrelated to or have no affect upon another study in which the researcher may be involved; such distinct findings require separate documentation).
- An IRB submission WILL NOT proceed to IRB review if the COI tab or COI “Status” that appears in your study workspace for any member of the study team is any of the following: (a) Team Member(s) without Certifications; (b) Awaiting Profile Update; and (c) Administrative Review (see image below).
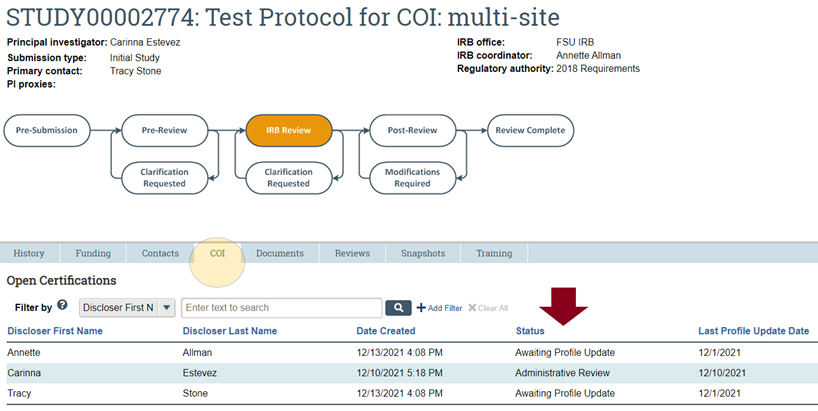
ANY of the above statuses pertaining to any member of a study team above will delay IRB review of a submission in the RAMP IRB queue. You are strongly advised to promptly tend to any CAMS submission for which one of the above statuses exists in order to avoid unnecessarily prolonging IRB review of your RAMP IRB submission.
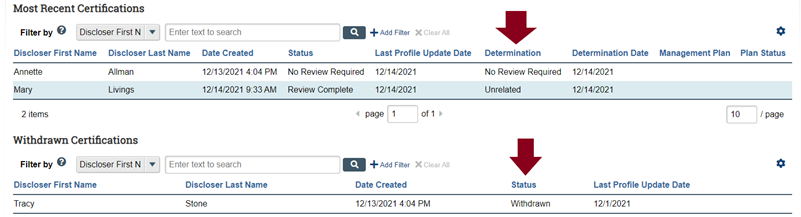
- An IRB submission may proceed to IRB review if the COI “Determination” or “Status” that appears in your study workspace for every member of the study team is one or more of the following: (a) No Review Required; (b) Unrelated; or (c) Withdrawn (see image below).
- If a determination is made that a member of a study team has a conflict of interest, direct and specific disclosure to research participants (human subjects) about such interests is required. Such disclosure must be included as part of the informed consent process, and may be conveyed as follows: in the first paragraph in the “What else do I need to know” section of the HRP-502 consent form, after the first paragraph in the “What is this study about” section of the HRP-502a or HRP-502c consent forms, or at the end of the first paragraph in the HRP-502i Information Sheet form. The IRB may require additional language. Refer to our HRP-502COI – TEMPLATE COI Consent Language Template in RAMP IRB (or here on the OHSP Templates & Required Forms web page; scroll down to the Consent-related Templates section and look for the Conflicts of Interest Consent Language Template) for different types of interests to disclose to human subjects. Any of these forms missing such language will be returned to the study team for correction.
- The IRB will not approve of an IRB submission (new, continuing review or modification) unless any related COI management plan is deemed by the IRB as having satisfied federal regulatory criteria for approval of research. This includes revisions to a COI management plan, as well as implementation of informed consent procedures through which study subjects are adequately informed about investigators’ conflicts of interest (see bullet above). Note that the IRB may regardless of any other FSU office’s conflict of interest review outcome determine that a conflict of interest does exist; require additional information about researchers’ interests; impose additional requirements in order to minimize such conflicts; require that study subjects be informed about such interests or conflicts; and take any other actions to ensure that human subjects are both protected from research risks implicated by a conflict of interest as well as informed about such conflicts of interest.*
- Submission in CAMS of any new or change in a COI disclosure for any study team member for an existing IRB study may, if a COI management plan is imposed based upon the submission, require a RAMP IRB study modification. If a study modification is not initiated by the PI and the OHSP/IRB becomes aware of the new or changed COI disclosure, the PI will be contacted to submit a study modification.
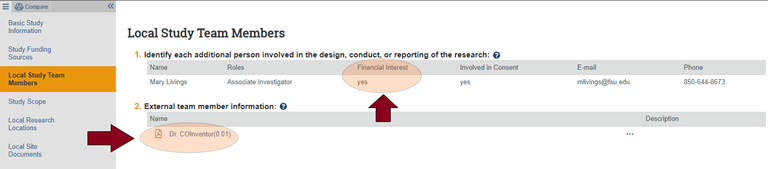
- COI disclosures reported in a RAMP IRB submission before implementation of the CAMS COI module will appear in the Basic Study Information section (Question 7) and/or Local Study Team Members section (Questions 1 and/or 2 for external team members) of your RAMP IRB SmartForm or workspace as depicted in the images below:
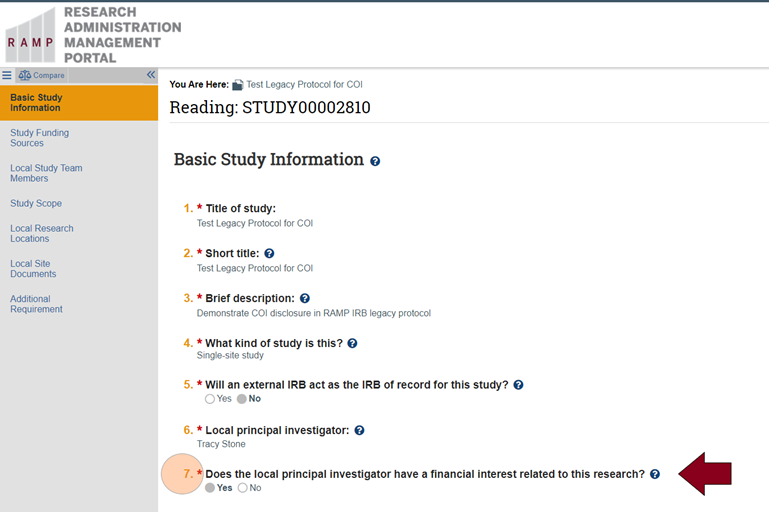
Note that the “?” button for Question 7 above provides related information and instructions as depicted below:
Review of these pre-CAMS RAMP IRB COI disclosures may be handled similarly as those that are reported in CAMS, including the requirement that human subjects be informed about the conflict of interest. Refer to our HRP-502COI – TEMPLATE COI Consent Language Template in RAMP IRB (or here on the OHSP Templates & Required Forms web page; scroll down to the Consent-related Templates section and look for the Conflicts of Interest Consent Language Template) for different types of interests to disclose to human subjects.
- Check out our Conflicts of Interest/CAMS web page for an overview, definitions, references and other helpful information.
- Authority for and support of CAMS and FSU conflicts of interest reporting requirements are provided by the FSU Office of Compliance and Ethics and the FSU Office of Research Compliance Programs, respectively, NOT the OHSP or IRB! For your questions or to obtain information about the CAMS COI system, including an overview, announcements, training, support and contacts, visit this CAMS project page.
- CAMS is accessible within the myFSU portal as an icon under the Links section, and you must therefore have a valid FSU user credential. The system is designed to be part of the single sign-on process, allowing FSU credentialed users to conveniently access CAMS along with other systems within a single platform using one set of login credentials. Use any of the means below to access the CAMS system to submit your study:
-
- Click on the CAMS icon within the myFSU portal; or
- Click on the CAMS link within an Outlook email notification (which occurs when you need to take action in the system).
- Type the CAMS website address in your internet browser: https://cams.fsu.edu
- To find out how to navigate through CAMS, click the "CAMS Quick Start Guide" or access the guide at this CAMS Project page, see the video "How to Update Your Disclosure Profile" or access the video in the COI/CAMS module Help Center, or submit a support ticket to the CAMS Support team.
IMPORTANT NOTICE:
Federal law provides the IRB with authority to, for example, review human research; require modifications to secure approval; and ensure that legally effective informed consent will be and is obtained, including disclosure of the above interests. FSU institutional officials may not approve of research involving human subjects that has not been approved by the IRB, including IRB approval of or required modifications for COI consent language (see 45 CFR §§46.109, 46.111(a)(4), 46.116; 42 CFR §50.605(a)(1)(ii); and 45 CFR §94.5(a)(1)(ii)).
- If you have a specific question not listed above, contact OHSP using the “Add Comment” feature (to the left of the workflow diagram and listed below under Next Steps) on your study workspace to ask your question and select the option to send an email notification about your question to your IRB Coordinator (whose name is listed above and to the left of the workflow); there, in response to the question about who should receive the email notification about your comment, select "IRB Coordinator." This will help to ensure that the IRB Coordinator assigned to your submission will receive a notification that your comment has been posted in your RAMP IRB workspace.
- Alternatively and if you have not yet submitted your study or your specific question is not related to your submitted study, contact OHSP or refer to the contact information in the left panel, and your question will be routed to OHSP staff for follow-up with you directly.
- For FAQs specific to student-led research, visit this student-led research FAQs page.
Need to contact OHSP? Click on the panel below.
Contact information for the IRB and OHSP are in the panel above. The garnet ribbon below is information for the FSU Office of Research or FSU generally, and does not include any information for the IRB or OHSP.