Clinical Trials
IRB-related guidance, information, instructions and other resources for the FSU research community

This Clinical Trials page provides the FSU research community with information about when a study may constitute a "clinical trial", researchers' additional responsibilities when planning or conducting clinical trials, and access to pertinent resources and other guidance. The information provided here is primarily focused upon matters with which the IRB has a special interest (i.e., ensuring that clinical trials conform to clinical trial-related human research regulatory requirements). Important note: federal funding agencies, other sponsors as well as regulatory agencies such as the FDA have additional requirements not specifically addressed here; questions and the need for information about those requirements should accordingly be redirected there. This page covers the following topics and more:
- When is a Study a Clinical Trial?
- What Do Researchers Need to Do?
- Sources of Regulatory Information
- Finding Related Resources
A subset of human research, clinical trials are subject to additional requirements imposed by different federal laws as well as study sponsors and publishers, due in part to the following: the use of regulated products such as drugs and devices; laws that mandate public access to information about clinical trials; heightened risks that are often associated with health-related interventions; and the involvement of study participants who may be vulnerable because of their poor health status and other factors. Knowing when your human research is a clinical trial for which additional requirements will apply may help to ensure appropriate IRB review of your studies, protection of study participants and ultimately compliance with applicable laws. Check out the panels below for more information.
For purposes of determining whether a study constitutes a "clinical trial" and may therefore be subject to clinical-trial related legal or other requirements, a threshold matter is ascertaining how a clinical trial is defined by the federal department or agency that may regulate or fund the study. Below are the definitions provided by the U.S. Food and Drug Administration (FDA), National Institutes of Health (NIH), and under the Federal Policy for the Protection of Human Subjects (aka the "Common Rule") that many federal departments and agencies have adopted.
- U.S. Food and Drug Administration (FDA) Definition of Applicable Clinical Trials (42 CFR Part 11; ClinicalTrials.gov)
Applicable clinical trials generally include:-
Controlled clinical investigations (other than phase 1 investigations) of any FDA-regulated drug or biological product for any disease or condition
-
Certain studies of FDA-regulated medical devices, excluding small clinical trials to determine feasibility and certain clinical trials to test prototype devices, but including FDA-required pediatric post-market surveillances of a device product
-
Interventional studies (with one or more arms) of FDA-regulated drug, biological, or device products that meet one of the following conditions:
-
The trial has one or more sites in the United States
-
The trial is conducted under an FDA investigational new drug application or investigational device exemption
-
The trial involves a drug, biological, or device product that is manufactured in the United States or its territories and is exported for research
-
-
- National Institutes of Health (NIH) Definition of Clinical Trial (NIH Policy)
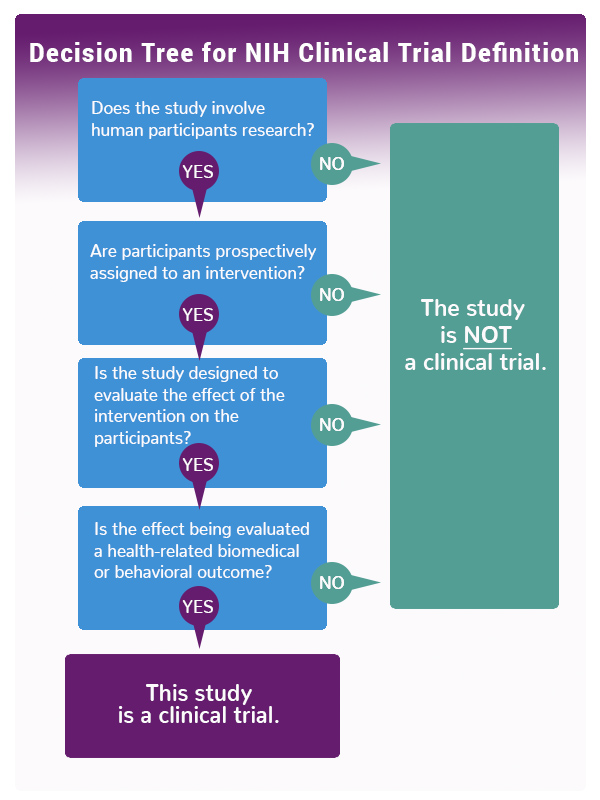
A clinical trial is a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes. If the answer is “yes” to all four of the following questions, then the research study meets the NIH definition of a clinical trial and must be registered:- Does the study involve human participants?
- Are the participants prospectively assigned to an intervention (placebo or control)?
- Is the study designed to evaluate the effect of the intervention on the participants?
- Is the effect being evaluated a health-related biomedical or behavioral outcome?
See the infographic below or use this NIH Decision Tool to see whether your study falls within NIH's definition of the term clinical trial.
-
Federal Policy for the Protection of Human Subjects (45 CFR Part 46, section 46.102(b))
A clinical trial means a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.
-
- Note that this definition may apply to studies that may or may not be subject to FDA regulations or funded by NIH, and which may be conducted or funded by other federal departments and agencies, non-federal sponsors and a wide range of research and academic institutions and organizations.
Infographic: NIH Decision Tree for Definition of a Clinical Trial
ClinicalTrials.gov is a web-based, publicly available resource about federally and privately supported clinical trials conducted in the United States and abroad, both current and past studies. This resource is intended to provide the public with ready access to information about clinical studies involving many different diseases and other health conditions. Researchers' registration and/or posting of certain clinical trial information to the web site is required if their study is funded by the NIH or another federal agency and/or subject to FDA regulation.
Clinical trial study information will include descriptions of the studies, copies of IRB-approved consent forms, study summary results and adverse event information, as may be applicable, for completed studies. The site requires that researchers and their institutions comply with certain requirements, such as updates and correcting errors. To learn more about ClinicalTrials.gov-related requirements for clinical trials studies, including background, the underlying laws, how to register studies, submit results, obtain training or FAQs, visit the ClinicalTrials.gov main web site or the ClinicalTrials.gov background page. A screenshot of the site is provided below.
Important Note! The OHSP/IRB does not provide support to researchers for their use, navigation or problems in using ClinicalTrials.gov. For FSU administrative assistance with ClinicalTrials.gov-related requirements, contact the FSU Clinical Research Hub. [link]
FAQs for ClinicalTrials.gov: Visit this page at https://clinicaltrials.gov/ct2/manage-recs/faq to obtain information and instructions for using the National Library of Medicine's ClinicalTrials.gov site. Numerous topics are covered, including how to register, deadlines, information and documents to submit, penalties for non-compliance and contacts. A snapshot of the site's background page and "Submit Studies" tab is depicted below.
 |