Revised Common Rule
On January 21st, 2019, the Florida State University IRB and Office for Human Subjects Protection (OHSP) started implementing the new requirements of the Revised Common Rule, as stipulated in the Federal Policy for the Protection of Human Subjects (referred to as the Common Rule since many federal agencies are signatories to the policy). The Revised Common Rule is the first major revision to federal policy, and is intended to facilitate research, remove ambiguity, reduce regulatory burden, and better protect human subjects involved in research. The most cited version of the Common Rule are federal regulations at Title 45 of the U.S. Code of Federal Regulations, Part 46.
Research may be subject to either the pre-2018 Requirements (“Common Rule”) or the 2018 Requirements (“Revised Common Rule”) versions of federal policy. More information about the applicability and transition provisions can be found within the Revised Common Rule at 45 CFR §46.101.
When Does the Revised Common Rule Apply?
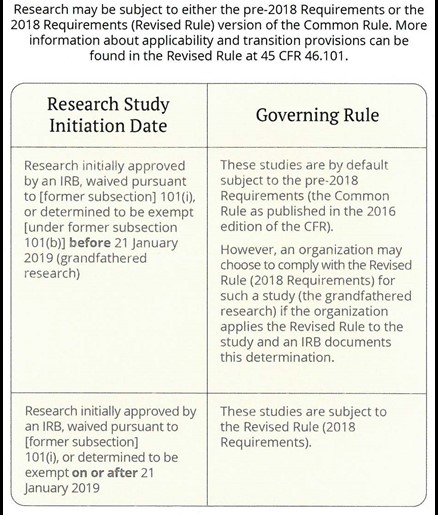
Source: CITI Program Final Rule Resources
Overview of Revised Rule Changes
Revised Human Subjects Research Definitions
- The definition of “human subject” has been changed to include “identifiable biospecimens” as well as how data and/or biospecimens have been collected or will be used.
- The definition of “research” has been expanded to list activities that are specifically deemed not to be research, i.e., scholarly or journalistic activities, including oral history, journalism, biography, literary criticism, legal research, and historical scholarship; public health surveillance; criminal justice or criminal investigative activities; and activities in support of intelligence, homeland security, defense, or other national security missions.
Other New Definitions
- Clinical trial – The old rule provided no definition of a clinical trial. The Revised rule defines a clinical trial as: “A research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of the interventions on biomedical or behavioral health-related outcomes.”
- Benign Behavioral Intervention – The old rule provided no definition of benign behavioral interventions. The revised rule defines a benign behavioral intervention as: “Brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on the subjects, and the investigator has no reason to think the subjects will find the interventions offensive or embarrassing.”
- Examples: having the subjects play an online game, having them solve puzzles under various noise conditions, or having them decide how to allocate a nominal amount of received cash between themselves and someone else.
New and Revised Exemption Categories
- Category #1: Revised to include a statement that the research cannot “adversely impact students’ opportunity to learn required educational content or the assessment of educators who provide instruction.”
- Category #2: Revised to include clarification that the data may involve visual or audio recording. For research that only includes interactions involving educational tests, survey or interview procedures, or observation of public behavior if at least one of the three provisions included in this exemption is met. This category also allows for the collection of sensitive, identifiable data to be collected as long as “limited review” is conducted by the IRB.
- New Category #3: Includes research with benign behavior interventions, i.e., interventions that are brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on the subjects; subjects not likely to perceive them as offensive or embarrassing. *Cannot involve deception unless deception is authorized by the participant. Limited IRB review is required if the information obtained is recorded by the investigator such that the identity of the subject can readily be ascertained either directly or through identifiers.
- Category #4: Revised to include mention of biospecimens and removes the pre-2018 requirement that information and biospecimens must be pre-existing.
- Category #5: Revised to require Federal department or agency conducting or supporting the research to publicly post research online (on a website to be specified by the federal government, or on clinicaltrials.gov) prior to commencement.
- Category #6: Unchanged (taste and food quality evaluation; consumer acceptance studies).
- New Category #7: Includes storage or maintenance of identifiable private information or identifiable biospecimens for potential secondary research use, for which broad consent is required. This exemption requires limited IRB review to determine that the requirements for broad consent are met; that broad consent is appropriately documented or documentation of broad consent is appropriately waived; and that there are adequate provisions in place to protect the privacy of subjects and maintain confidentiality of the data, if there will be a change made for research purposes in the way the identifiable private information or identifiable biospecimens are stored or maintained.
- New Category #8: Includes research involving the use of identifiable private information or identifiable biospecimens for secondary research use, for which broad consent is required. This exemption requires an IRB to determine through limited review that there are adequate provisions in place to protect the privacy of subjects and maintain confidentiality of the data, and that the research to be conducted is within the scope of the obtained broad consent.
[Refer to §46.104(d)(2)(iii), §46.104(d)(3)(i)(C), §46.104(d)(7), and §46.104(d)(8)(iii) of the revised Common Rule.]
Exemption and Vulnerable Populations
- Pregnant Women – Pregnant women are no longer considered a “vulnerable population.” All exemptions may apply if the condition of the exemption is met.
- Prisoners – None of the exemptions apply, except for research aimed at involving a broader subject population and only incidentally includes prisoners.
- Children – Exemptions (d)(1) and (d)(4-8) may involve children. Exempt (d)(2) (research on educational tests, surveys, interviews or observations) may include children, but:
- Exempt (2)(i) and (ii) studies are permitted but ONLY if the studies involve educational tests or observation of public behavior when the investigator(s) do not participate in the activities being observed; and
- Exempt (2)(iii) is not applicable to research with children. This exemption is where the investigator can readily ascertain the identity of the child.
Limited Review
Limited review is a new process introduced by the Revised Rule that does not require an IRB to consider all of the approval criteria in §46.111 for certain exemptions. This was designed to ensure adequate privacy safeguards for identifiable private information and identifiable biospecimens. Limited review involves the IRB making and documenting a determination that certain conditions, specified in the regulations, are met to ensure adequate provisions are in place for protecting privacy and maintaining confidentiality. Limited review may be done via an expedited review process and may be required for exemption categories 2, 3, 7, and 8.
[Refer to §46.109(a) and §46.109(f)(1)(ii) of the revised Common Rule.]
Broad Consent
Broad consent is a new term introduced by the Revised Rule that allows an investigator to seek prospective consent to unspecified future research. It’s intended for “secondary research use” of private identifiable information and identifiable biospecimens. Broad consent is required for exempt categories 7 and 8. The FSU IRB has decided to consider Broad Consent on a case-by-case basis.
[Refer to §46.116(d) of the revised Common Rule.]
Continuing Review
For research initially approved on or after January 21st, 2019, continuing review is no longer required under the following circumstances*:
- Research eligible for expedited review.
- Research reviewed by the IRB in accordance with the limited IRB review.
- Research that has progressed to the point that it only involves (a) data analysis, including analysis of identifiable private information or identifiable biospecimens, and/or (b) accessing follow-up clinical data from procedures that subjects would undergo as part of clinical care.
Investigators are still responsible for submitting modifications for review/approval prior to implementation; reporting new information within 5 business days, and closing human research protocols, as appropriate.
(*IMPORTANT NOTE: The OHSP can override this default and still choose to require continuing review, as long as the IRB documents the decision and the rationale for this decision.)
[Refer to §46.109(f), §46.110, and §46.115(a)(8) of the revised Common Rule.]
Informed Consent Changes
Please always check our Templates and Required Forms for updated consent form templates.
According to the Revised Rule, the consent process must first include a presentation of “key information,” i.e., information most likely to assist in understanding why to participate (or not) in the research within an introductory paragraph. As per the Revised Rule preamble, key information must include the following:
- The fact that consent is being sought for research and that participation is voluntary
- The purpose(s), the expected duration of participation, and the procedures to be followed
- The reasonably foreseeable risks or discomforts to the prospective subject
- The benefits to subjects or others that may reasonably be expected
- Appropriate alternatives, if any, that might be advantageous
NOTE: If information included in the "key information" section also satisfies the elements of informed consent under §46.116(b) and (c), this information need not be repeated later in the body of the informed consent.
[Refer to §46.116(a) of the revised Common Rule.]
New Basic and Additional Elements of Consent
- If the research involves the collection of identifiable private information or identifiable biospecimens, a statement on whether the identifiers might be removed and information or biospecimens could be used for future research without additional informed consent.
- When applicable:
- A statement that biospecimens (even if identifiers are removed) may be used for commercial profit and whether the subject will or will not share in this commercial profit.
- A statement about whether clinically relevant research results, including individual research results, will be disclosed to subjects.
- A statement about whether the research project might include whole genome sequencing.
[Refer to §46.116(a) of the revised Common Rule.]
Single IRB Review
When more than one institution is involved in a research study, the regulations define this as a “cooperative research project.” The Revised Rule requires the use of one IRB for cooperative research (“Single IRB” or “sIRB”).
NOTE: The effective date for this change was January 19, 2020; the NIH Policy on sIRB, which requires a single IRB for NIH-funded human research, went into effect January 25, 2018.
Additional Resources
- Office for Human Research Protections (OHRP)
https://www.hhs.gov/ohrp/education-and-outreach/revised-common-rule/revised-common-rule-q-and-a/
- Public Responsibility in Medicine & Research (PRIM&R)
https://www.primr.org/commonrule/resources-tools/
https://www.primr.org/commonrule/regulatory/
- CITI Program Final Rule Resources
https://about.citiprogram.org/en/final-rule-resources/
- HRP Consulting Group
https://thehrpconsultinggroup.com/2017/01/19/new-final-common-rule-summary-of-changes/
- Revised Common Rule (45 CFR 46)
